Gladstone NOW: The Campaign Join Us on the Journey✕


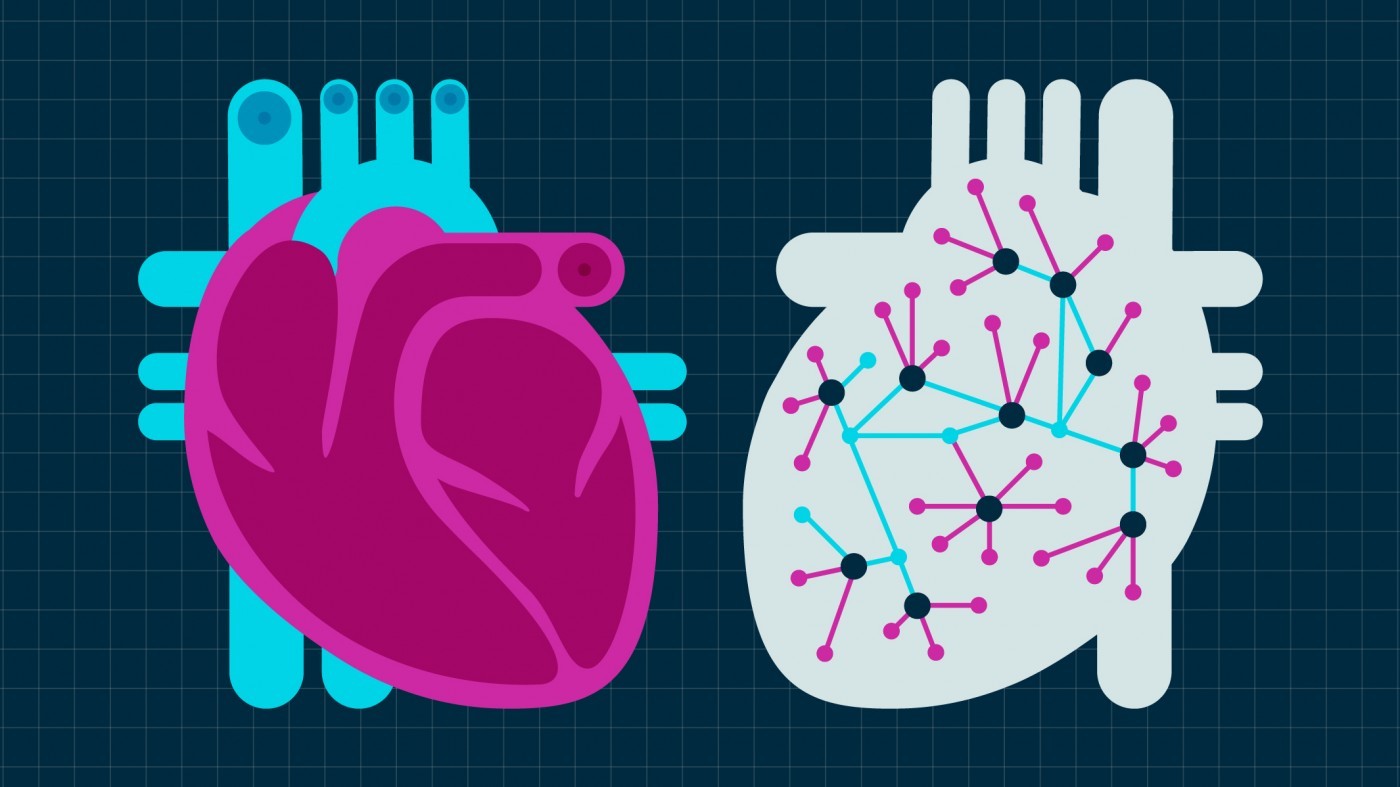
Gladstone scientists are teaming up to understand and overcome heart disease. With a battery of modern technologies including iPS cells, CRISPR-based genome editing, whole-genome sequencing, mathematical modeling, and machine learning, they are unravelling the networks of signals, cells, and proteins that build a strong beating heart, and how they go awry in congenital heart defects and other heart diseases.
It takes a precise combination of cells and signals at exactly the right time in the development of a fetus to make a strong, beating human heart. Alone, one type of cell can model some functions of the heart in a laboratory culture dish, but will never pump blood through a living organism. And just as growing a heart takes cellular, genetic and molecular teamwork, the study of cardiovascular disease is equally multifaceted. It takes the right cadre of researchers—multidisciplinary, collaborative and creative—to understand the heart’s complexity, what can go awry in the vital organ, and how to respond when that happens.
At Gladstone Institutes, teamwork among scientists happens every day. And it is revealing new insight into cardiovascular disease.
“We have a critical mass of experts in different disciplines, which is allowing really creative discovery in this space,” says Gladstone President Deepak Srivastava, MD.
Fittingly, their collaborative work focuses on partnerships: Which proteins bind to each other inside heart cells? Which combinations of genetic mutations cause disease? How do different cell types interact to make a heart?
“We have individuals who are not shy about coming together to ask these big questions,” says Benoit Bruneau, PhD, director of the Gladstone Institute of Cardiovascular Disease.
Medleys of Mutations
More than a decade ago, Srivastava, who is a pediatric cardiologist, encountered a family with two seemingly healthy parents yet two children were born with heart defects and a third was stillborn with a similar heart problem. He suspected that each parent must carry genetic mutations that don’t cause disease alone but—when inherited in combination—affect heart formation. At the time though, genetic sequencing was expensive and sequencing each family member’s DNA would likely not be enough to confirm what was causing disease.
“Even if you know the inheritance patterns and have a family’s genome sequences, everybody has a lot of genetic variation,” says Srivastava. “Sifting through that to find the meaningful variations is challenging.”
Then came faster and cheaper sequencing, along with the advent of stem cell technologies developed by Gladstone Senior Investigator, Shinya Yamanaka, MD, PhD, recipient of the 2012 Nobel Prize for his discovery of induced pluripotent stem (iPS) cells. Similar to embryonic stem cells, these cells have the potential to become nearly every cell in the body, including heart cells.
Srivastava and his team took advantage of these technological advances to resume studying the family he’d met years before. In collaboration with Gladstone Senior Investigator Bruce Conklin, MD, they coaxed iPS cells generated from the patients’ skin cells into beating heart muscle cells, thus generating a source of cells to study the family’s disease.
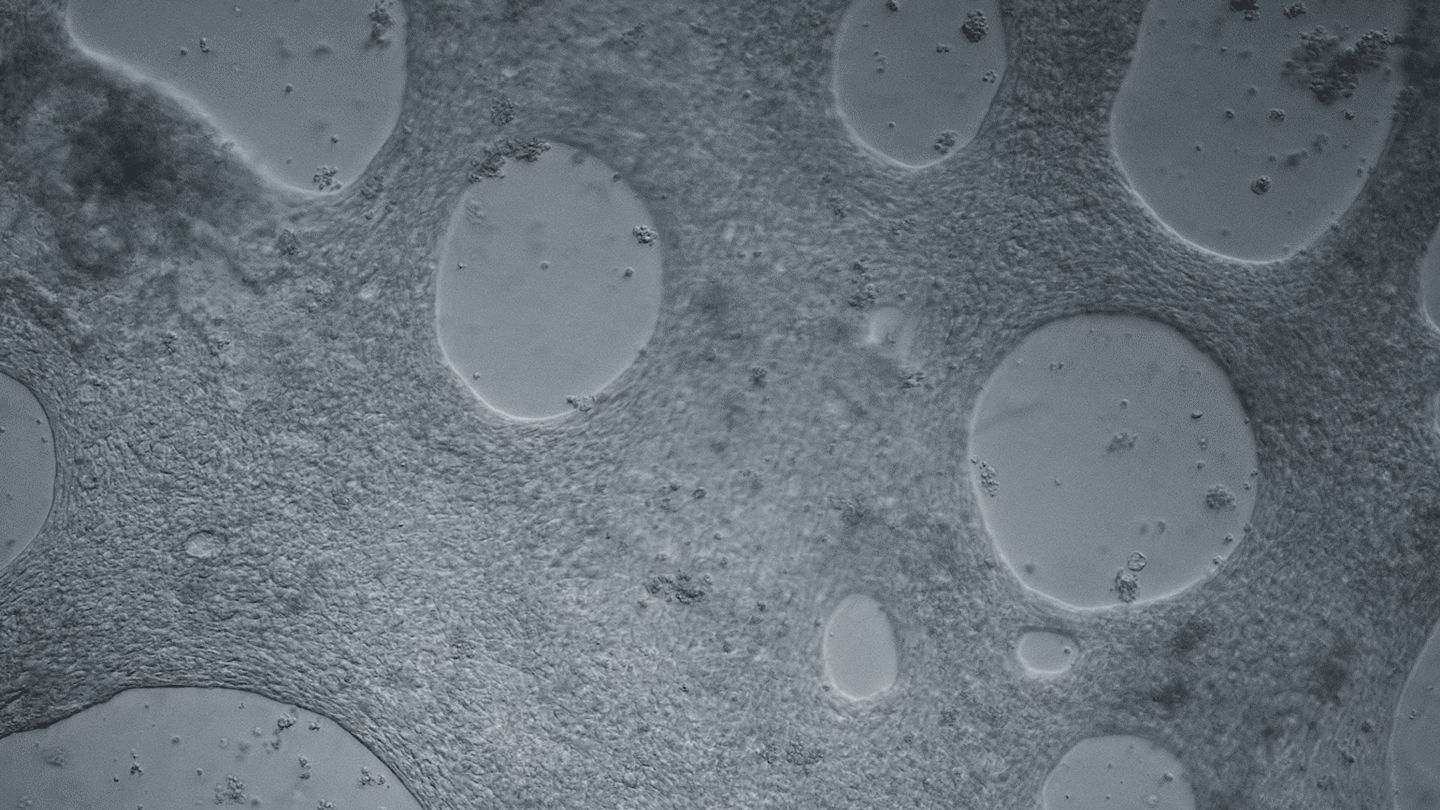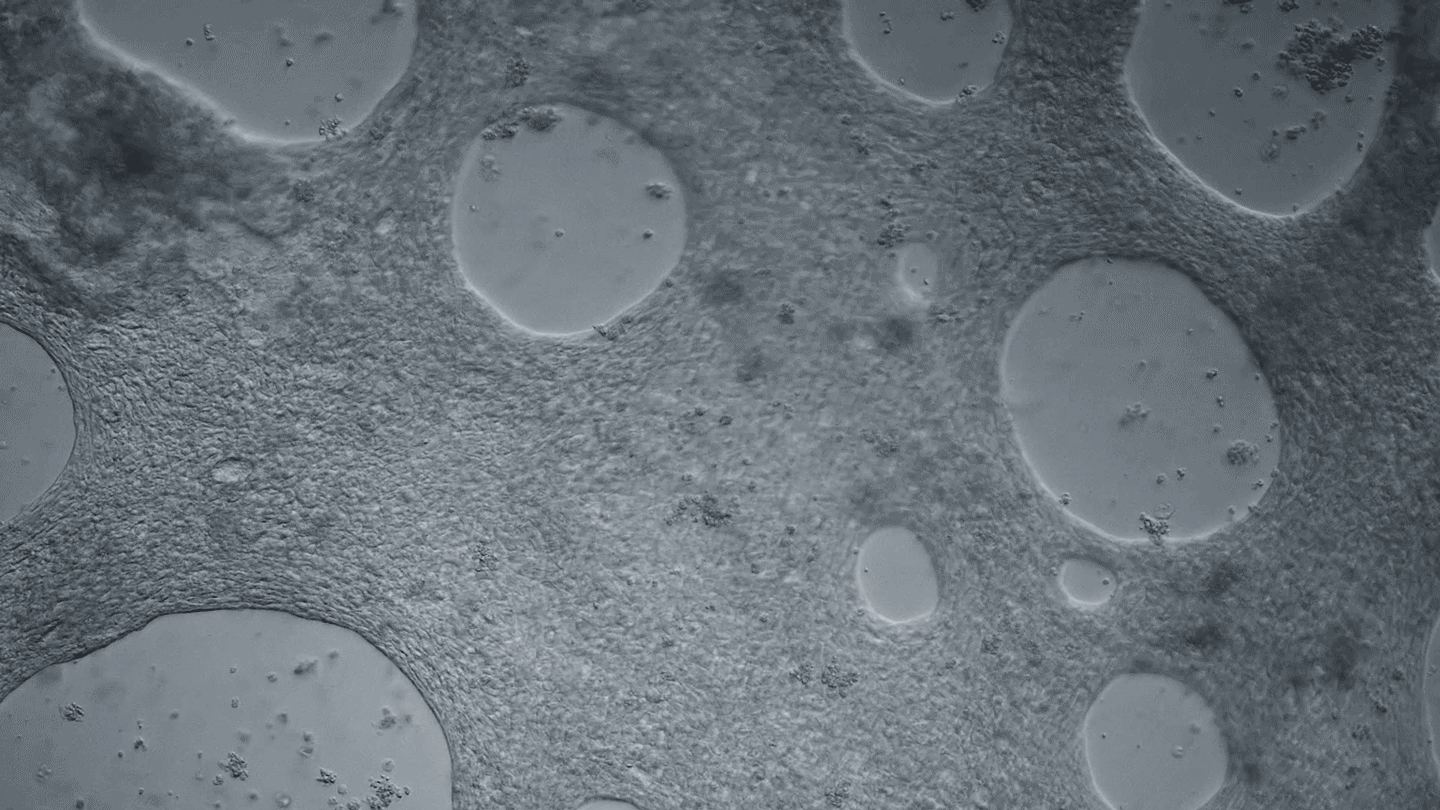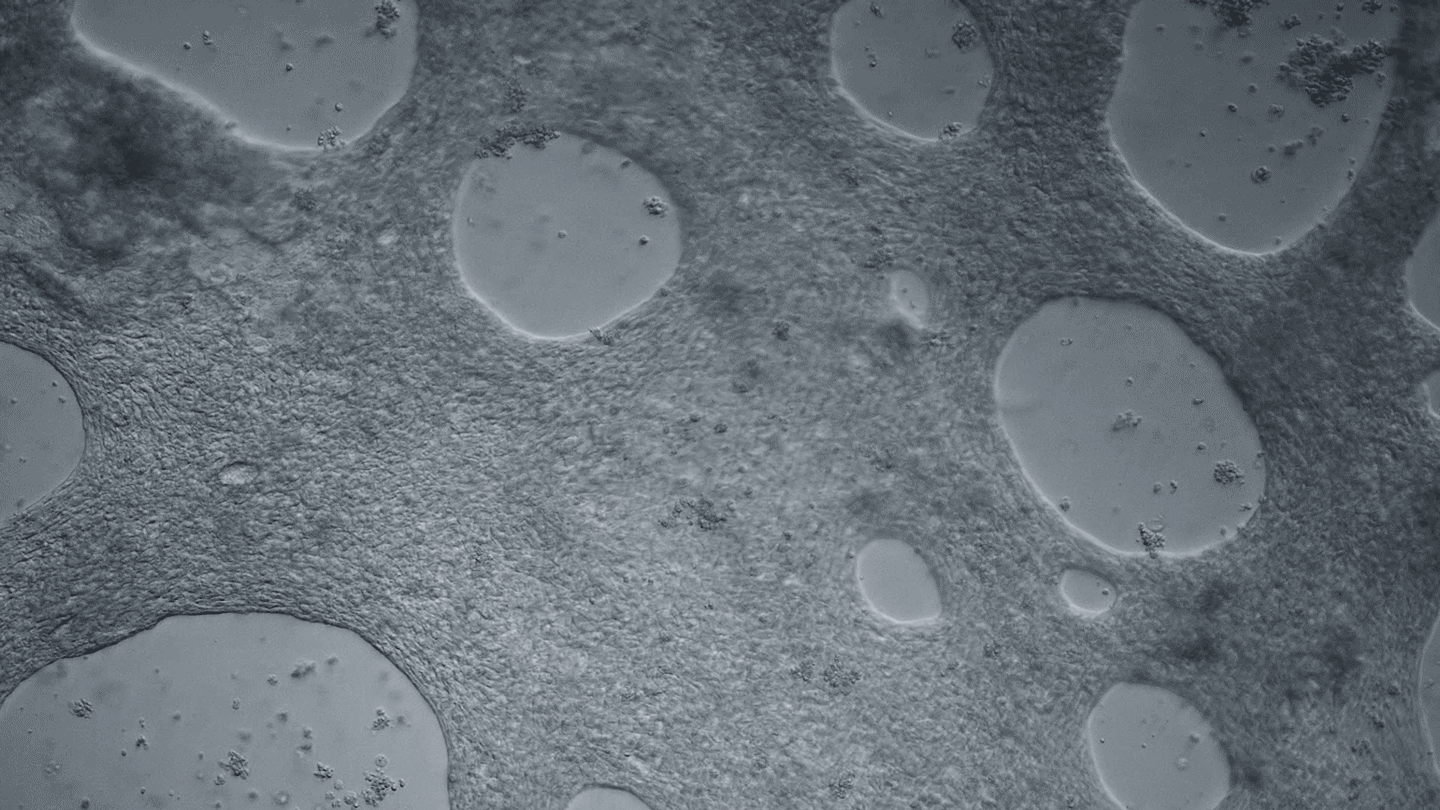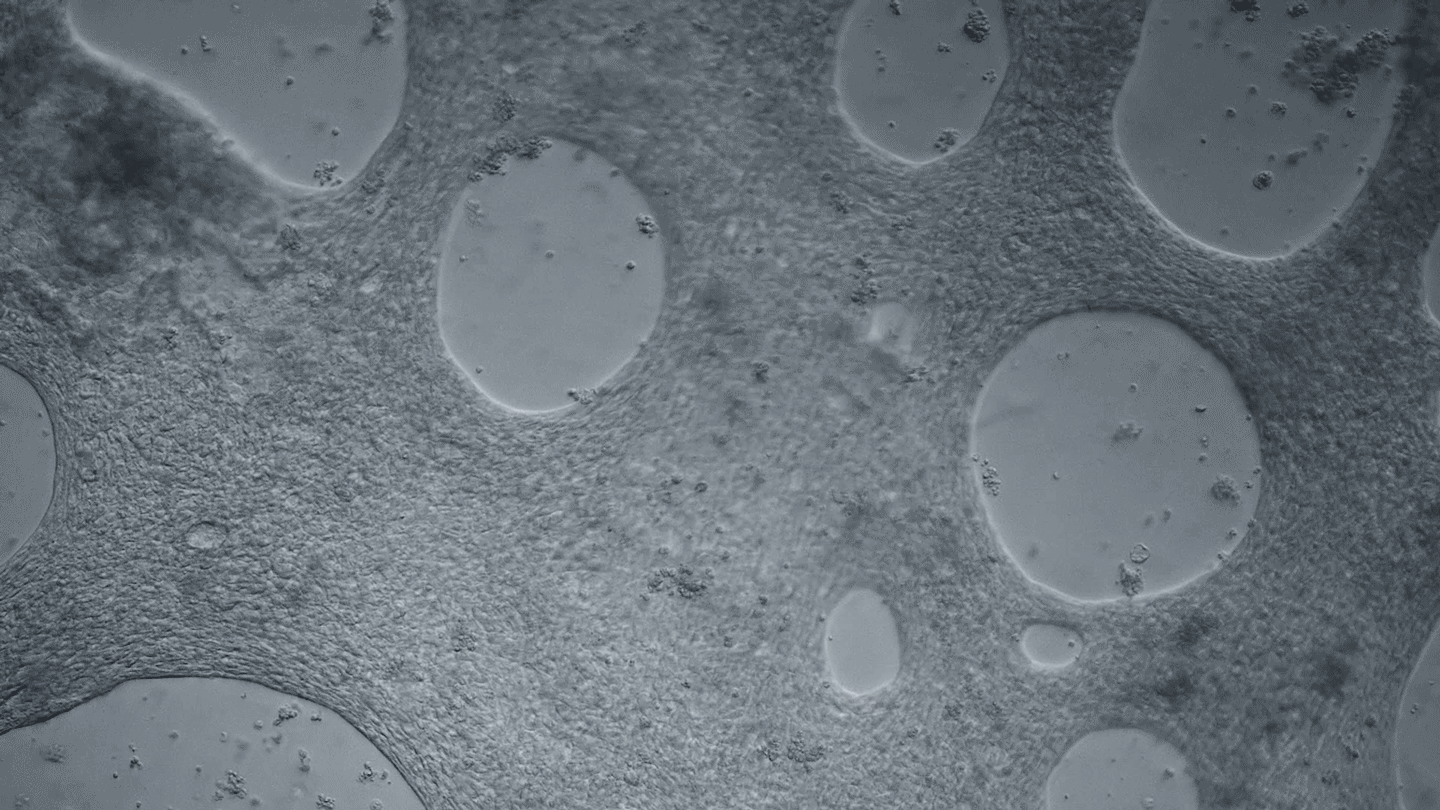
Heart cells derived from human iPS cells produce tissue-like sheets that beat synchronously. Video by Juan A. Pérez-Bermejo.
Srivastava and his colleagues also sequenced the genomes of family members and discovered that the father had two genetic mutations associated with an increased risk of heart disease. The mother had a third mutation that hadn’t been previously associated with disease. Applying new CRISPR gene-editing tools developed by Gladstone Senior Investigator Jennifer Doudna, PhD, to mice and iPS models, the researchers confirmed that the three mutations, when combined, caused disease.
Encouraged by this discovery, Srivastava—along with Gladstone collaborators including Katie Pollard, PhD, director of the Gladstone Institute of Data Science and Biotechnology—is mining genomic databases of people born with congenital heart disease, and their parents, to uncover other detrimental combinations of gene variants.
To help narrow down the list of possibilities, Pollard’s team will rely on a computational model of heart development that they have fine-tuned through collaborations with Gladstone researchers who, over the years, have accumulated much knowledge about how the heart forms. Their model links together this knowledge about the cells, genes, and proteins that contribute to heart development.
“We’ve spent a lot of time studying the fundamental blueprint for making a heart,” says Pollard. “And the motivation for doing that is that we want to understand how mutations can break that process.”
Once the model makes a prediction about the effect of a mutation, that prediction can be tested in the lab. Srivastava’s lab will use CRISPR-based technologies to test the consequences of thousands of different combinations of mutations in human iPS cells. Running these experiments in parallel, using technology that allows scientists to analyze single cells, the lab will study which combinations affect the development of the stem cells into heart cells.
Protein Partners
In addition to revealing the cause of heart defects in patients, pinpointing genetic mutations can shed light on how different molecules work together in heart cells. If a combination of mutations causes disease, it’s likely because the proteins they encode are part of the same cluster of molecules—either physically or functionally.
So Pollard is teaming up with Nevan Krogan, PhD, another Gladstone senior investigator, to home in on proteins that pair up inside heart cells, using a technique known as affinity purification mass spectrometry. The researchers isolate a protein of interest, along with anything that happens to be attached to it at the time, then characterize all the proteins that came along for the ride.
“Often in biology we study things just one at a time,” says Pollard. “But we know that inside cells, proteins don’t operate alone; they work in networks.”

Senior Investigators Katie Pollard, Benoit Bruneau, and Deepak Srivastava are uncovering networks of proteins and genes controlling heart cell formation in health and disease.
Bruneau’s group has studied one such protein network in depth. TBX5 was one of the first genes ever associated with congenital heart disease; children with mutations in this gene can be born with a variety of heart and limb malformations. The TBX5 protein acts as a transcription factor—it controls the expression of dozens of other genes. While a mutation in a typical gene only affects the protein encoded by that gene, a mutation in a transcription factor can have farther-reaching effects.
To discover what other genes TBX5 controls, Bruneau’s team generated iPS cells with missing or mutated versions of TBX5 and differentiated the cells into heart cells. Then, they measured the expression levels of every gene in the genome, to discover which were affected by TBX5. If a gene is controlled by TBX5, it might produce less RNA when TBX5 is absent or mutated.
“We were able to computationally build a gene regulatory network that shows us connections between TBX5 and other genes that it interacts with,” explains Bruneau.
More work is needed to understand these kinds of networks, though, and relate the findings back to patients with heart defects.
“It’s a slow road,” says Bruneau. “But a wonderful aspect is that when we discover a new layer of complexity, it opens up a whole set of other questions that we didn’t know we even needed to ask in the first place. And that’s what I find really exciting.”
Answering those questions, he hopes, will lead to new therapeutic avenues, such as drugs that can target some of the molecular partners of TBX5.
Learning from Healthy Hearts
Bruce Conklin too has focused on protein interactions to better understand mutations related to heart disease. People with a particular mutation in a gene called BAG3 are less likely than the rest of the population to have heart failure or need a heart transplant, and their hearts pump blood more robustly than average. Conklin was curious why this was, so his lab undertook a series of experiments to study how mutations in the BAG3 protein changed its interactions with other proteins.
Conklin’s group carried out their experiments in heart cells derived from iPS cells with the normal BAG3 and the protective version.
“In cardiac cells we saw a big change in which proteins BAG3 interacted with,” recalls Conklin.
Conklin’s team discovered that many of the proteins that interact with BAG3 in heart cells are related to how cells respond to stress. The protective version of BAG3, they showed, ramps up this stress response, letting cardiac cells better cope with a heart attack, a toxin, or a pathogen.
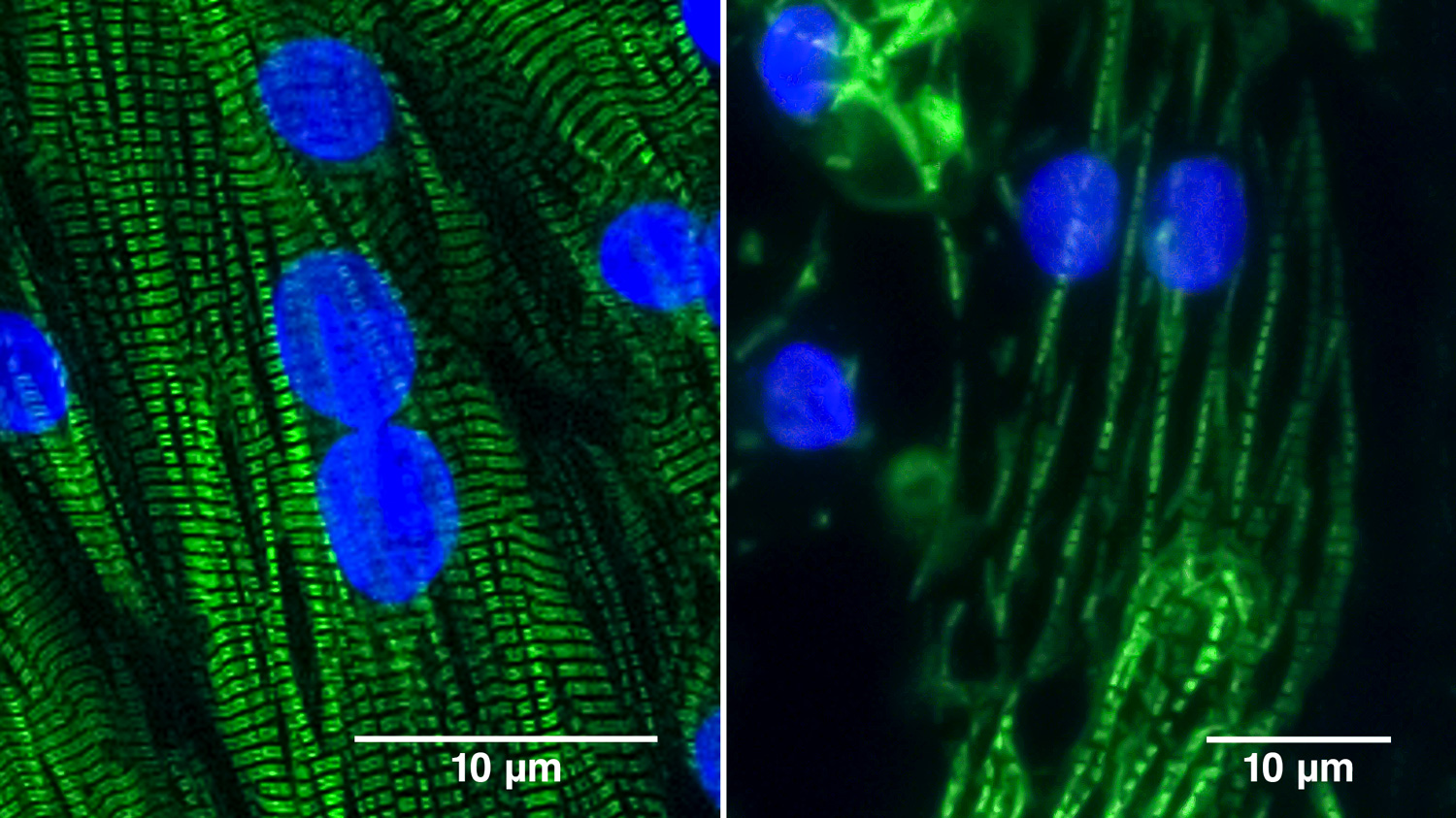
Heart muscle cells derived from normal human iPS cells (left) contain well-organized sarcomeres (green), the structures that contract and allow the cells to beat. Heart muscle cells lacking the BAG3 protein (right) form broken and disorganized sarcomeres. The cells’ nuclei appear in blue. Photos by Juan A. Pérez-Bermejo.
To understand BAG3 even better, Conklin and his colleagues are mutating each of its newly discovered partners-in-crime and studying the effect on heart cells. But identifying small changes in how heart cells look by eye is difficult. So the team turned to a machine-learning algorithm; the computer program could analyze pictures of thousands of heart cells and pinpoint which ones looked physically different than the rest.
“By eye, we couldn’t always tell the difference between cells that harbored mutations and those that didn’t. But the machine-learning program saw differences we couldn’t,” says Conklin.
The program let them narrow down the list of BAG3’s binding partners to a handful of proteins that are most critical to heart cell formation. The researchers next plan to investigate whether any chemical compound can mimic the effect of BAG3—protecting people from heart failure by ramping up the stress response pathway in cardiac cells.
It’s not just proteins that communicate with each other: regions of DNA also interact, and these interactions determine what genes turn on and off in cells and when. With a new $13 million Program Project Grant from the National Institute of Health, Bruneau, Conklin, Krogan, Pollard, Srivastava, and their UC San Francisco collaborator Brian Black, PhD, will study how the three-dimensional structure of DNA inside heart cells dictates their development and health.
Outside Influences
Most research by Gladstone investigators on cardiovascular disease relies heavily on stem cell technology. Differentiating stem cells into heart cells shows scientists how the cells develop in an embryo, lets them create isolated heart cells from patients with disease, and offers a platform to study both the underlying cause of disease and possible treatments. But most of these experiments are done by growing cells in a flat culture dish—a far reach from a beating heart. Cells growing into three-dimensional clusters, many researchers think, might be a better proxy for what happens in an embryo.
“Right now, we can make heart cells aggregate into a three-dimensional structure that has some features of a tissue,” says Gladstone Senior Investigator Todd McDevitt, PhD. “But if you compare it to a section of a real heart, it doesn’t look that close.”

Senior Investigators Bruce Conklin and Todd McDevitt use iPS cell–derived cardiac cells to model heart development and understand genetic mutations that lead to heart diseases.
McDevitt wants to change that—his research aims to better understand the molecular cues that guide heart cell differentiation during embryonic development. Mimicking those cues could not only lead to more realistic heart tissue in the lab, but new ways to regenerate damaged heart tissue in people. In recent years, for instance, his lab studied the role of the extracellular matrix—a web of molecules that surrounds heart cells and fills the spaces between them.
During embryonic development, the heart maintains the ability to repair damage and regenerate tissue. After birth, this regenerative capacity is gone. McDevitt’s group compared the matrix surrounding fetal and adult heart cells in mice and humans, and found several molecules that differ between the two stages. Changing levels of these molecules, many of which relate to scar formation, may help coax adult heart tissue to scar less and generate new cells.
“The idea is that if we can find small molecules to modulate the matrix, that might actually help with tissue regeneration,” says McDevitt.
His team has also found that other neighboring cells help dictate the development of heart muscle cells—or cardiomyocytes. Researchers had already observed that mixing support cells called fibroblasts with cardiomyocytes helps the cardiomyocytes form more stable tissue. McDevitt’s group has now uncovered the exact molecular effect of fibroblasts on cardiomyocytes. This finding could help scientists refine the methods they use to generate heart tissue from stem cells. Moreover, the resulting tissue can be used to better mimic a functioning heart in studies of the effects of mutations, such as those ongoing in Srivastava’s and Bruneau’s labs.
“If we want to model diseases or do drug screening, we need to have adult cardiomyocytes that closely resemble those in the body,” says Bruneau.
Bench to Bedside
The questions being asked by Gladstone researchers revolve around the complex processes it takes to make a healthy heart. Their ultimate goals, however, include not only satisfying scientific curiosity but helping prevent and treat heart disease.
By the time most congenital heart defects are diagnosed—whether that comes when a baby is in utero, after birth, or late in life—it’s too late to turn back the clock on heart development. A structural abnormality must be corrected surgically. Over the last decades, improvements in such surgeries have meant many times more children born with such defects survive into adulthood. But that’s not always the end of the story.
“These kids often grow up to have other heart-related ailments later in life,” says Bruneau.
In some cases, those ailments—heart failure or disturbances in the heart’s rhythm—result from the same genetic mutations that led to the initial structural defect. So, learning how to target those genes or related molecular pathways can help prevent heart problems in these patients as they get older.
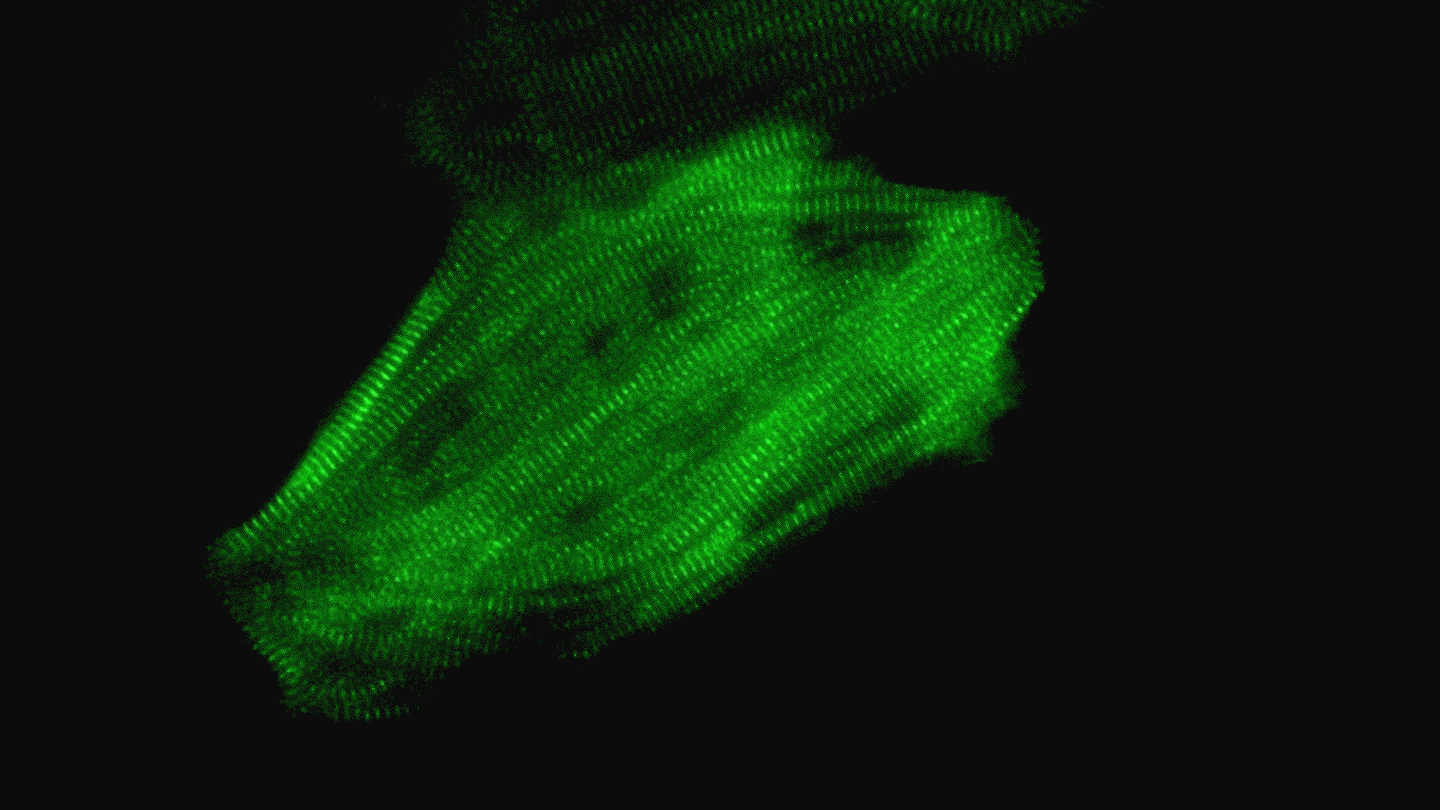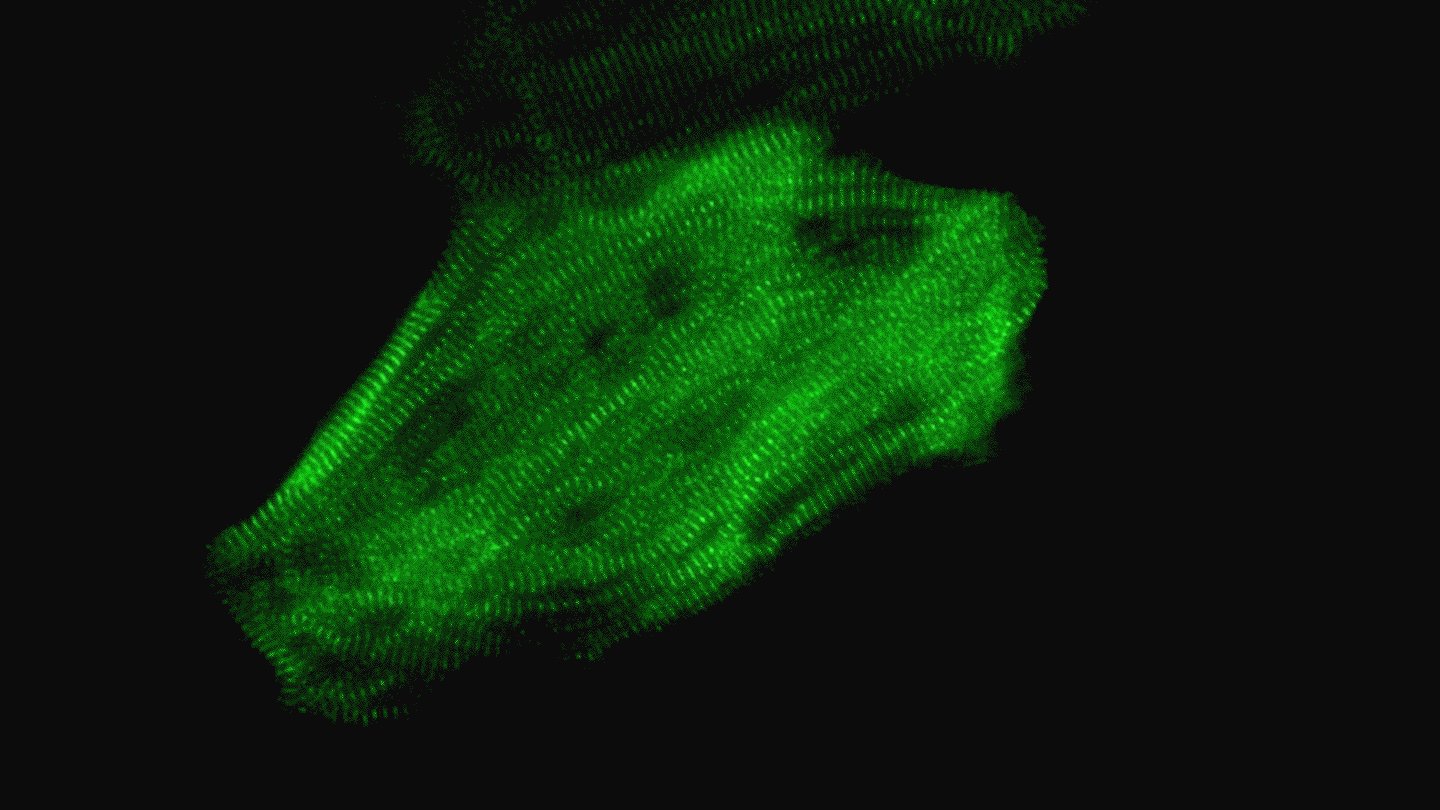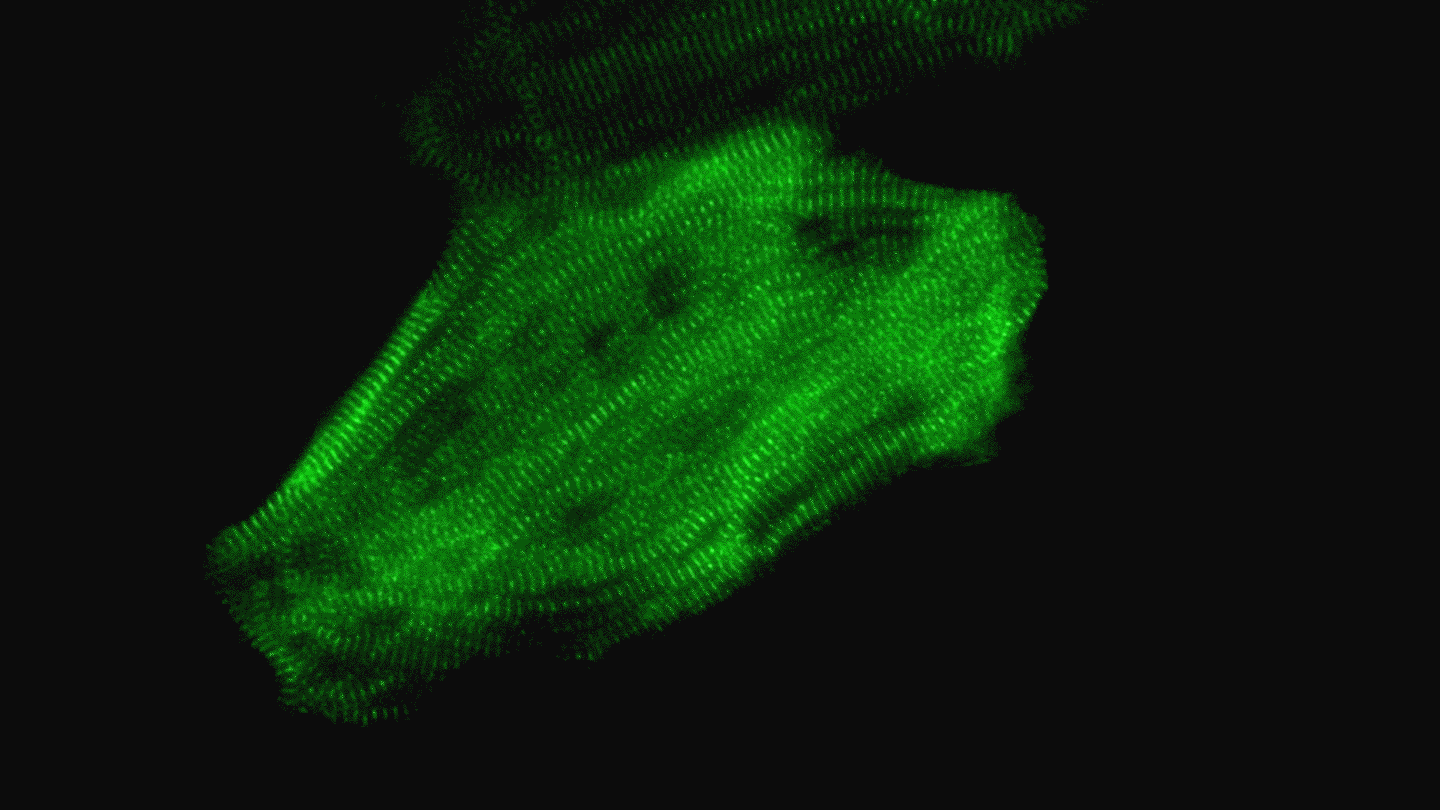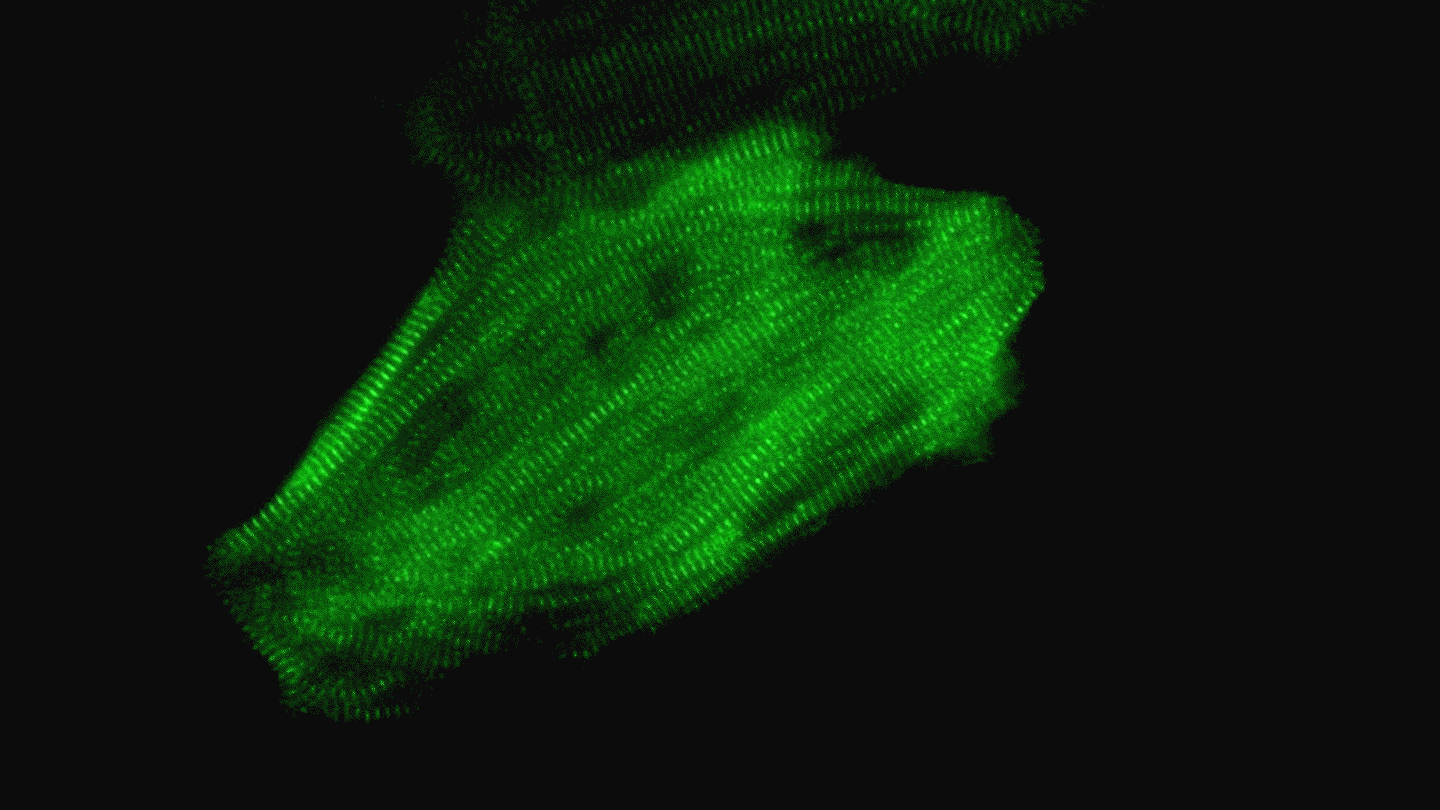
A heart muscle cell derived from a human iPS cell beats regularly thanks to a well-organized array of contracting structures known as sarcomeres (green). Video by Juan A. Pérez-Bermejo.
But the potential of the growing knowledge base is even bigger.
“The longer-term hope and dream is that we can find a way to actually prevent heart disease from occurring, particularly in children born to parents who are genetically susceptible,” says Srivastava.
He imagines something like folic acid—recommended to all pregnant women to lower the risk of spina bifida in developing fetuses. A compound that helps ensure that key molecules are present at the right levels in the developing heart could be similarly broadly prescribed.
Such a preventive, however, is still years away, and there may never be a single drug that can target all congenital heart defects.
“We realize there’s probably not going to be just one magic bullet that fixes these kinds of things,” says McDevitt. “The heart can go wrong in so many ways.”
And of course, the vast majority of cardiovascular diseases today are adult-onset conditions related to high blood pressure and blocked arteries; the risk factors are not only genetic but related to diet and lifestyle. According to the World Health Organization, more people around the globe die from such cardiovascular disease each year than die from any other cause.
Research into the molecular pathways that coax stem cells to differentiate into heart cells or give regenerative capacity to heart cells could one day lead to new therapeutics that help stave off heart failure in people at risk, or repair the scar tissue that results from a heart attack. Already, Gladstone researchers have shown how to transform scar-forming cells in the heart into new beating heart cells, regenerating damaged hearts from within.
If this approach—promisingly successful so far in animal models—could be applied to living people, it might reverse the damage to the heart that ensues from a heart attack. Already, this technology has been spun out into a start-up company in South San Francisco, Tenaya Therapeutics, that is developing a gene therapy product to test in people in the near future.
But for all these approaches, continued team work is needed.
“Together we synergize very strongly,” says Bruneau. “A single lab can’t do all the things that we need to overcome heart disease. But if we all come together and participate in understanding mechanisms underlying cardiovascular disease, we will be able to alter the course of what remains humankind’s biggest killer.”
Support Discovery Science
Your gift to Gladstone will allow our researchers to pursue high-quality science, focus on disease, and train the next generation of scientific thought leaders.
Turning the Tide on Tau: Q&A With Gladstone’s Lennart Mucke

Article
July 1, 2026
Turning the Tide on Tau: Q&A With Gladstone’s Lennart Mucke
Nearly 20 years after his landmark study uncovered novel roles of tau in Alzheimer’s disease, Gladstone's Lennart Mucke shares his perspective on new clinical data that could transform the future of brain health.
Mucke Lab Alzheimer’s Disease Gladstone Experts Neurological Disease News ReleaseThis Is AI Decoding the Genetic Cause of Disease

Video
March 23, 2026
This Is AI Decoding the Genetic Cause of Disease
In this video, Katie Pollard and Deepak Srivastava explain how Gladstone scientists are combining AI models with novel tools in the lab to finally decode the entire human genome.
Gladstone Experts Pollard Lab Srivastava Lab AIScience in Seconds | A New Path Toward Life Without Daily HIV Pills

Video
March 20, 2026
Science in Seconds | A New Path Toward Life Without Daily HIV Pills
In this video, Nadia Roan and Ashley George explain how they uncovered a new path toward long-term health without the need for daily HIV pills.
Gladstone Experts Research (Publication) HIV/AIDS Infectious Disease Roan Lab